TPJ March 2023 Editor choice: A Common Resequencing-Based Genetic Marker Dataset for Global Maize Diversity
A Common Resequencing-Based Genetic Marker Dataset for Global Maize Diversity
Grzybowski et al.
Embracing diversity – A genetic marker dataset with increased marker density facilitates association studies in maize
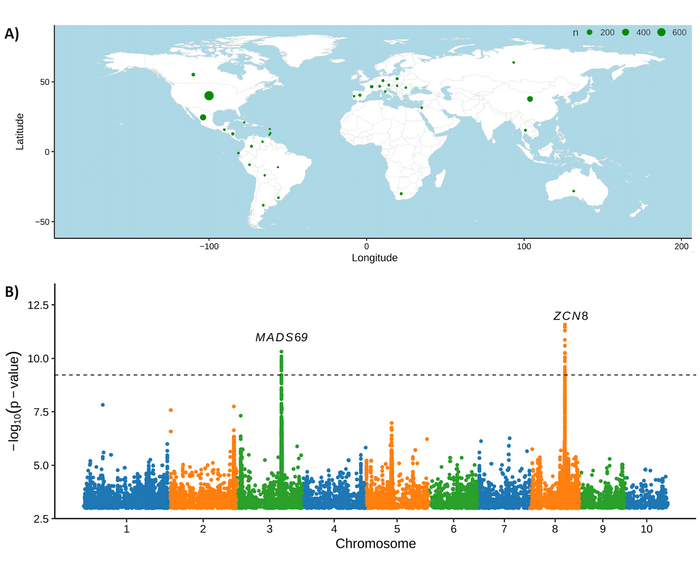
Figure 1: Marker dataset for global maize diversity can be used to identify new genes associated with a trait.
A) Geographical distribution of the countries of origin for the 1515 maize individuals used in this study.
B) Association test using the marker set defined in this study identified MADS69 and ZCN8 based on female flowering data (days to silking). Figure adapted from (Grzybowski et al., 2023).
Maize is known for its phenotypic and genetic diversity. On average, two maize lines diverge from one another as much as humans do from chimpanzees. This diversity is attributed to pollen flow between domesticated maize and its wild relative teosinte, and to trading between farmers. Maize diversity contributes to its adaptability to new climates and growth conditions, so that nowadays maize is grown across a wider area than any other crop.
Analysing maize populations based on DNA sequence polymorphisms (markers) is the premise for identifying selection targets and understanding geographic relations. Many studies revealed changes in genetic diversity by re-sequencing maize lines with differing sequencing depth, and with samples from across America and Europe. However, comparing markers between datasets of different studies can be challenging: datasets can differ in allele frequencies, in the SNP-calling pipelines, or in stochastic distributions of read depths, and therefore might identify different markers even in the same genomic region. Therefore, Marcin Grzybowski, James Schnable, and their team set out to unify previous datasets as well as incorporate newly re-sequenced samples in one dataset.
For the diversity dataset, Grzybowski et al. used whole genome resequencing data from 1,515 maize samples, comprising lines from the Wisconsin Diversity panel, inbred lines from Poland, as well as wild relatives, tropical landraces, archaeological samples, and modern open-pollinated varieties. Overall these samples originated in or were developed over six continents (Figure 1A). The authors identified over 350 million potential DNA sequence polymorphisms, second-stage quality filtering of the dataset reduced the number of variants to ∼46 million higher confidence variants.
Population genetic analyses are based on DNA sequence diversity, but also require the analysis of phenotypic traits. However, comparing phenotypes across different environments adds more variance and thereby reduces the statistical power to link genotype and phenotype. Comparing genotypes within the same environment is desirable, but not all genotypes are adapted to the same environments and can complete their life cycle. To tackle this problem, researchers use association panels, which maximize genetic diversity by selecting genotypes adapted to a specific environment. The authors used their marker set to analyse the Wisconsin Diversity Panel, and found that the lines retained over 70% DNA sequence variation compared to the wild relative Zea mays ssp. parviglumis, suggesting that there is still a lot of genetic variation in this diversity panel. Therefore, the marker dataset can serve as a resource for other researchers to calculate accurate values of DNA sequence diversity for their populations.
To analyse the impact of the high marker density on the outcomes for genome-wide association studies (GWAS), the authors used a published set of female flowering data generated from temperate-adapted maize inbreds. A previous GWAS using around 400.000 RNAseq-based markers identified the flowering time gene MADS69. With the newly generated marker set, the authors identified both MADS69 and a new locus, ZCN8, a gene that contributes to maize adaption to temperate climates (Figure 1B). The authors speculate that ZCN8 was previously not discovered because the dataset used RNAseq-based genetic markers and therefore missed significant SNPs in the intergenic space. The authors are hopeful that the higher density of markers will also help to achieve more precise localization of the causal variants associated with specific GWAS hits. Because of the diversity range of the maize lines used, this dataset can also be used to detect selection patterns in the genome associated with traits of interest, for example those related to domestication, adaptation to the environment, or genetic improvement during breeding.



